ScarfWeb supports powerful, fast and easy analytics of single-cell genomics data.
What is an analysis in ScarfWeb?
An analysis run will perform the following tasks:
- Cell filtering: Remove poor quality cells using multiple parameters, as we will see in the step 3 below
- Data normalization: Scarf will automatically perform normalization of count data using, cell size normalization for RNA modality, CLR normalization of the ADTs (Antibody derived tags) and TF-IDF normalization for ATAC modality.
- Feature selection: Not all the genes are equally useful for
Loading Analysis page
- To setup an a analysis run for a dataset. Click on the
Analyzebutton next to the name of the dataset. This button will appear if the dataset is ready to be analyzed and when it has not been analyzed before.
- When a dataset has been analyzed already and you would like to reanalyze it, then click on the “three dots" icon indicated below and then click on `Reanalyze`.
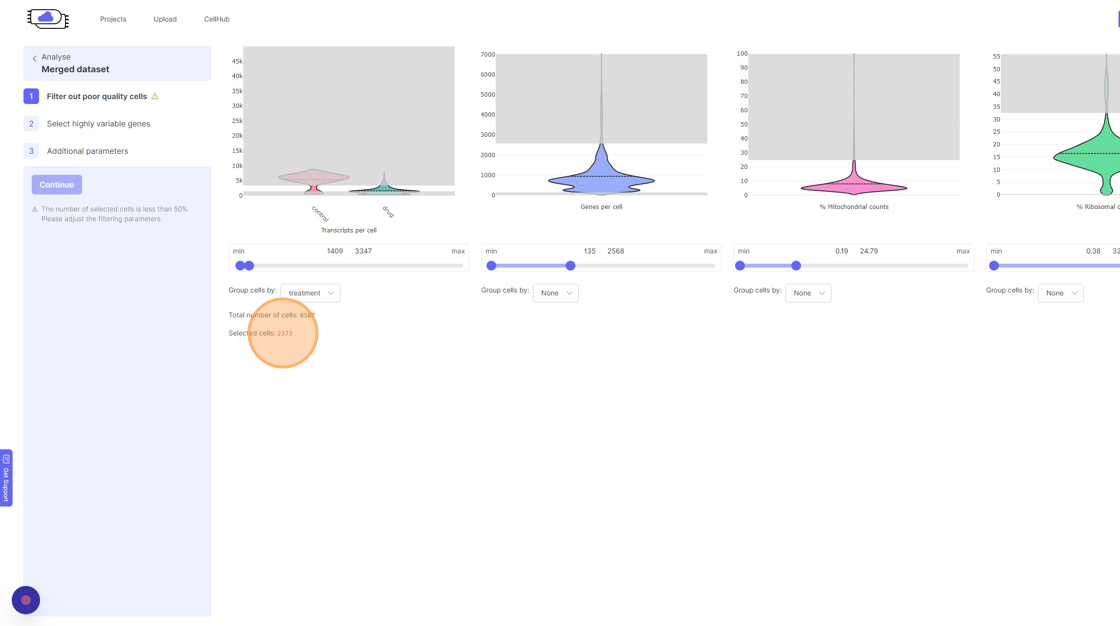
Cell filtering on Analyze Page
Now on the
Analyze page. You will see the three steps of the analysis setup. The first step is “Filter out poor quality cells”. In this step you will notice four violin plots on the right side. - Violin plots: They capture the distribution of a given value/metric (as listed below) across all the cells. The bulged part of the violin plot indicates the value for most of the cells and the dotted line across the violin plot indicates the mean value. The line (or the thin part of the violin plot) that extends out of violins plot indicates the outliers. These violin plots visualize four important quality control metrics for cells:
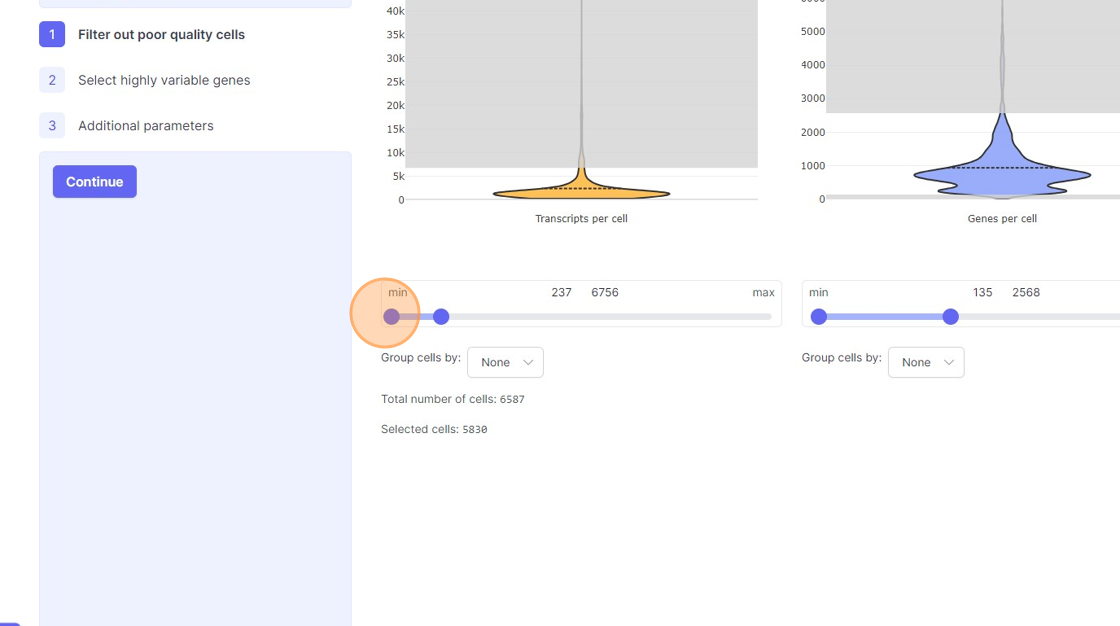
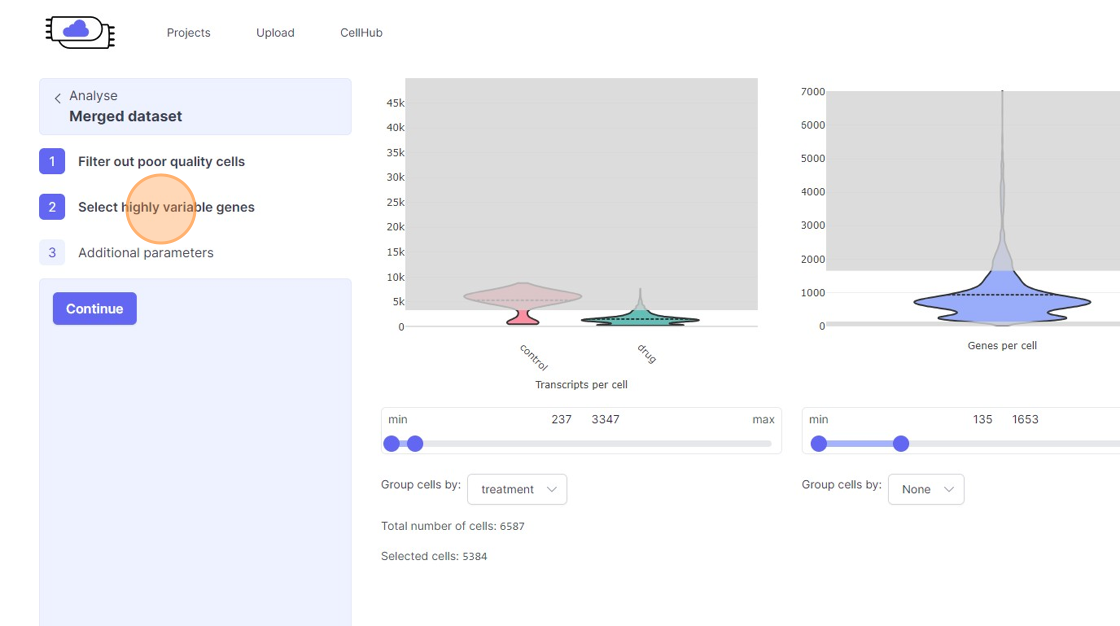
- Transcripts per cell: This is the total sum of all the observed reads/UMIs for each cell. One should remove the cells that are outliers on the higher side of the distribution. On the lower side of the violin plot, if there is secondary bulge then that can indicate low quality cells or non-cell barcodes that should be filtered out. Suggestions for filtering: Usually, having less than 500 transcripts in a cell can make the data very susceptible to noise and one risks including non-cell barcodes. Another rule-of-thumb that can be used during the filtering is to not have more than 20x difference between the lower and higher threshold. So, for example, if your lower threshold is 1000, then the higher threshold should ideally be less than 20000. A large difference in the upper and lower threshold can exacerbate technical artifacts, fi any, and limit the utility of data normalization. However, this is a general indication and there might be biological reasons that justifies not following this rule.
- Genes per cell: Here we summarize the total number of “detected” genes per cell. A gene is considered detected if there is at least one read/UMI attributed to that gene in a given cell. This is an essential quality control metric and varies a lot depending on the underlying single-cell library preparation method used. For example, droplet-based and split-pool combinatorial indexing based techniques tend to detect lower genes per cell compared to Smart-Seq and Quartz-Seq. Suggestions for filtering: Having less than 200 transcripts in a cell can make the data very susceptible to noise and one risks including non-cell barcodes into the analysis. Having a large difference in the upper and lower threshold values here can have an even more dramatic effect than transcripts per cell.
- % Mitochondrial counts: Here the percentage of all the reads/UMIs from a cell that originate from mitochondrial genes are shown. Cells with high % mitochondrial reads can be indicative of poor quality cell capture. However, there might be cases due to underlying biological reasons wherein a cell has a large number of mitochn
- % Ribosomal counts:
- Remove the outliers on the higher end of the distribution by moving the right side handle of the slider towards left.
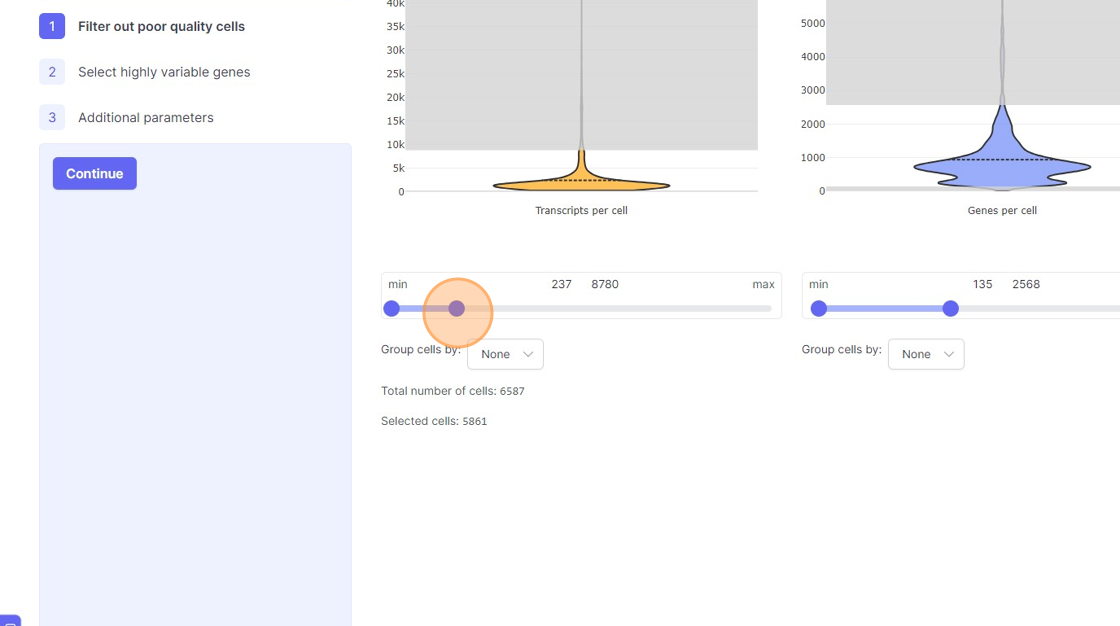
- Use the left part of the slider and move it towards the right to remove the cells from the lower end of the distribution.
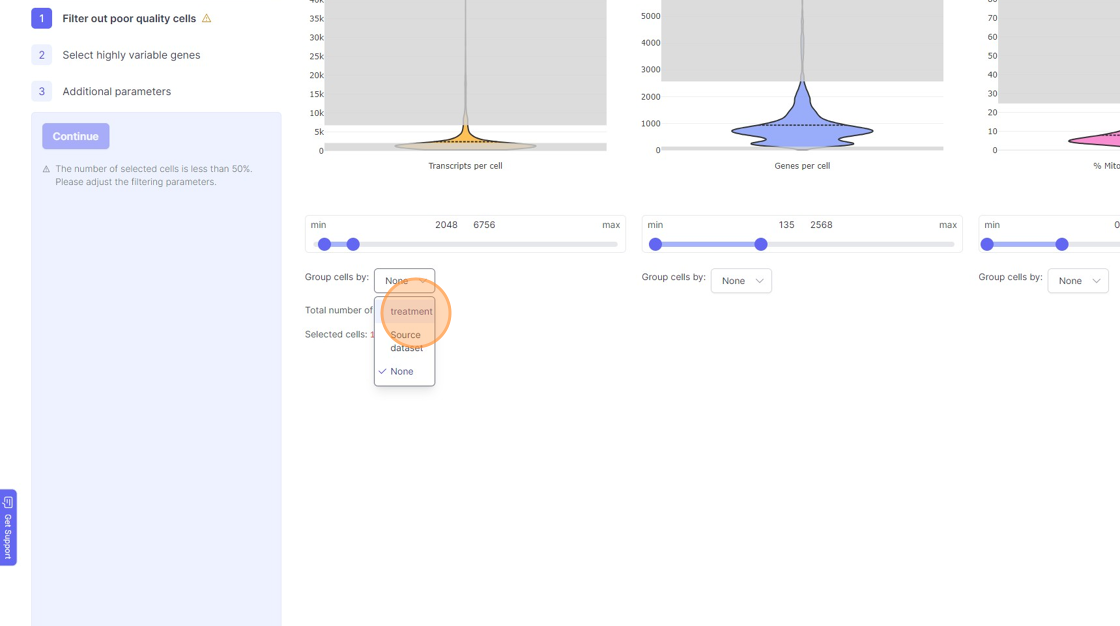
- If you are running a reanalysis or have have merged together samples, then you may have certain categorical metadata available in the “Group cells by” option below the violin plot for each of the filtering category.
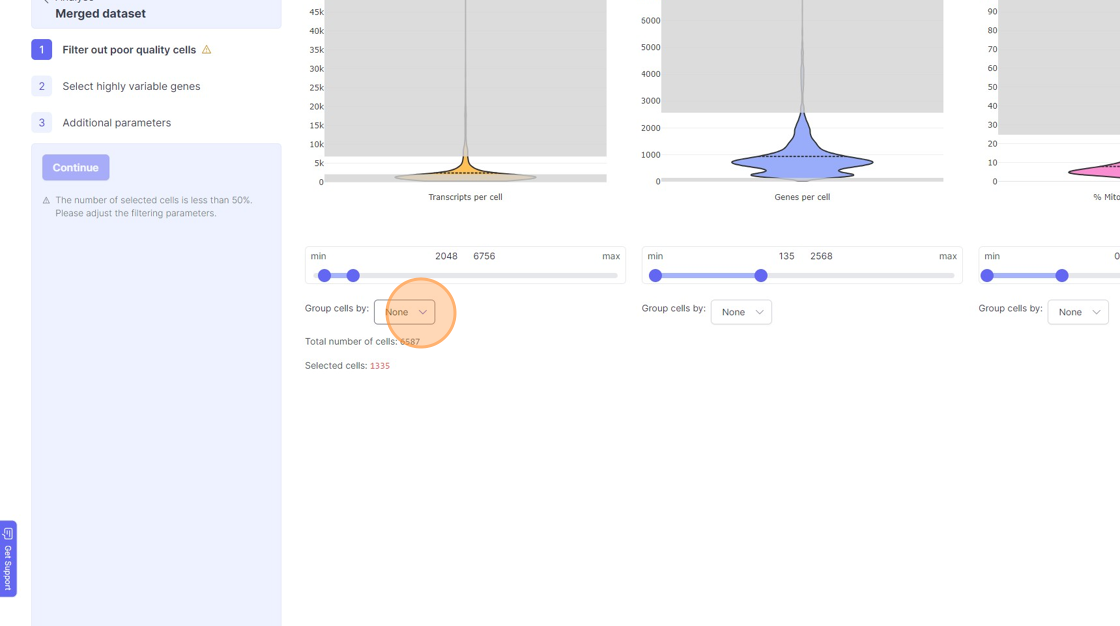
11\. Click "treatment"
Highly Variable Gene selection
30\. Click here.
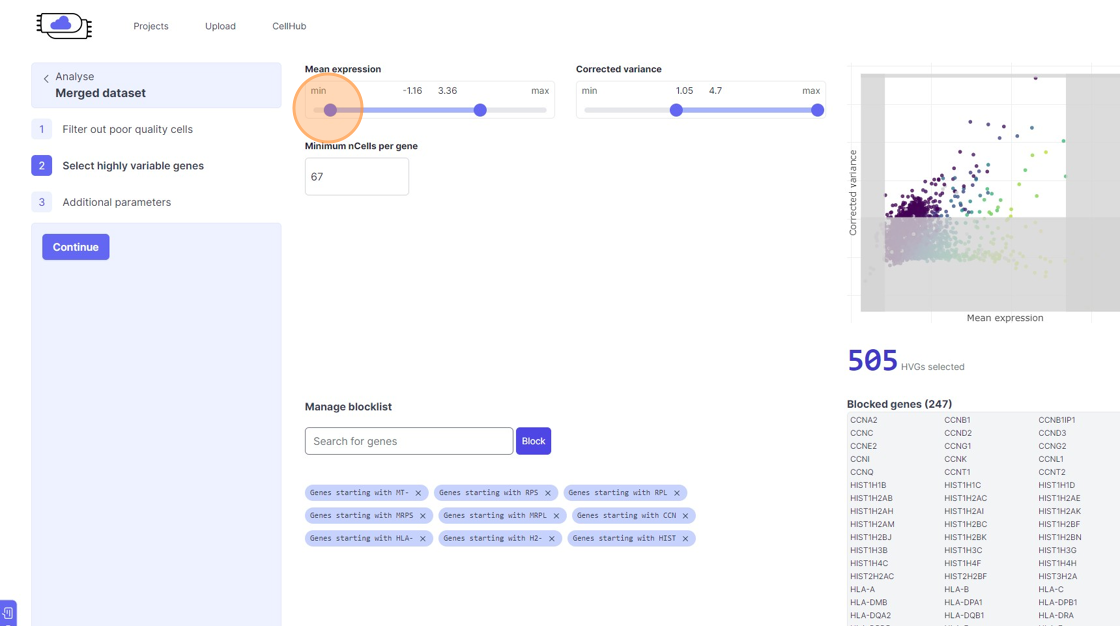
31\. Click here.
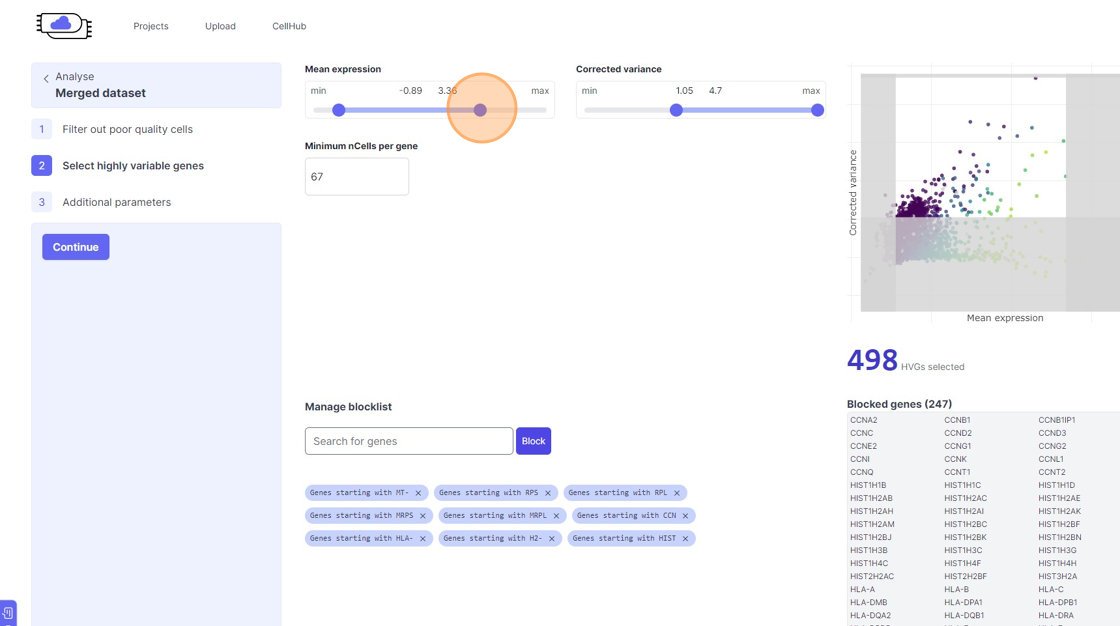
32\. Click here.
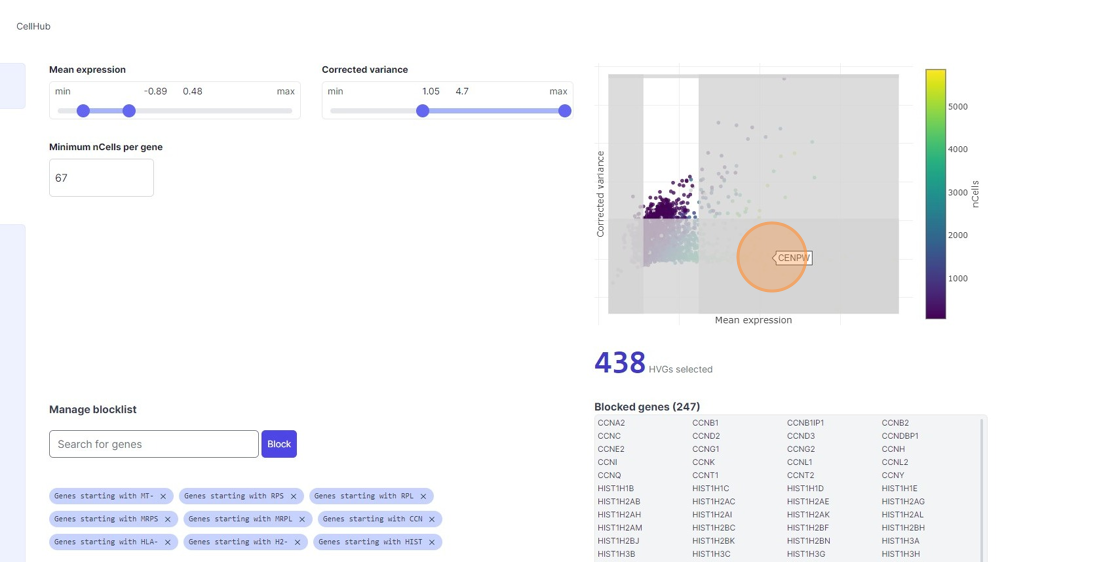
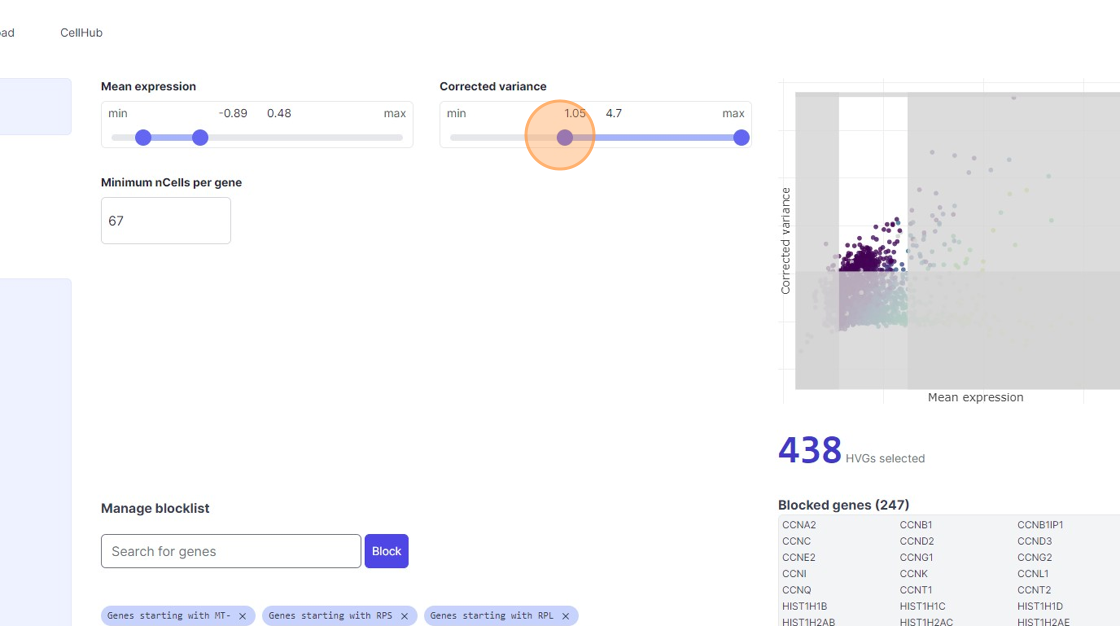
33\. Click here.
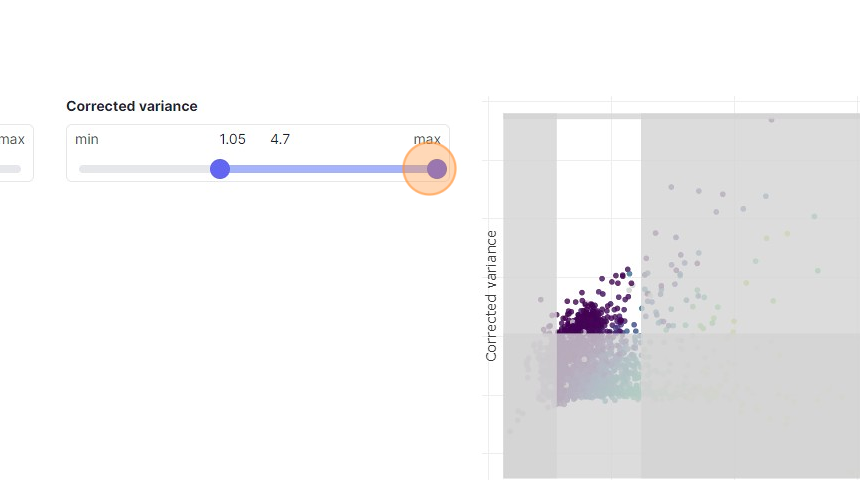
34\. Click here.
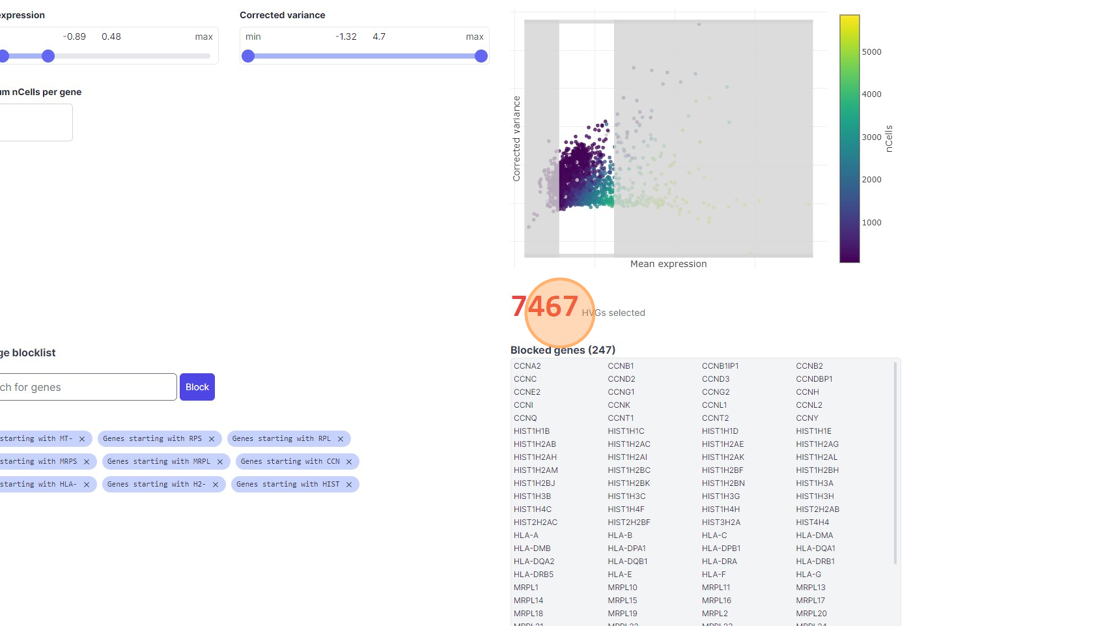
35\. Click "7467"
36\. Click here.
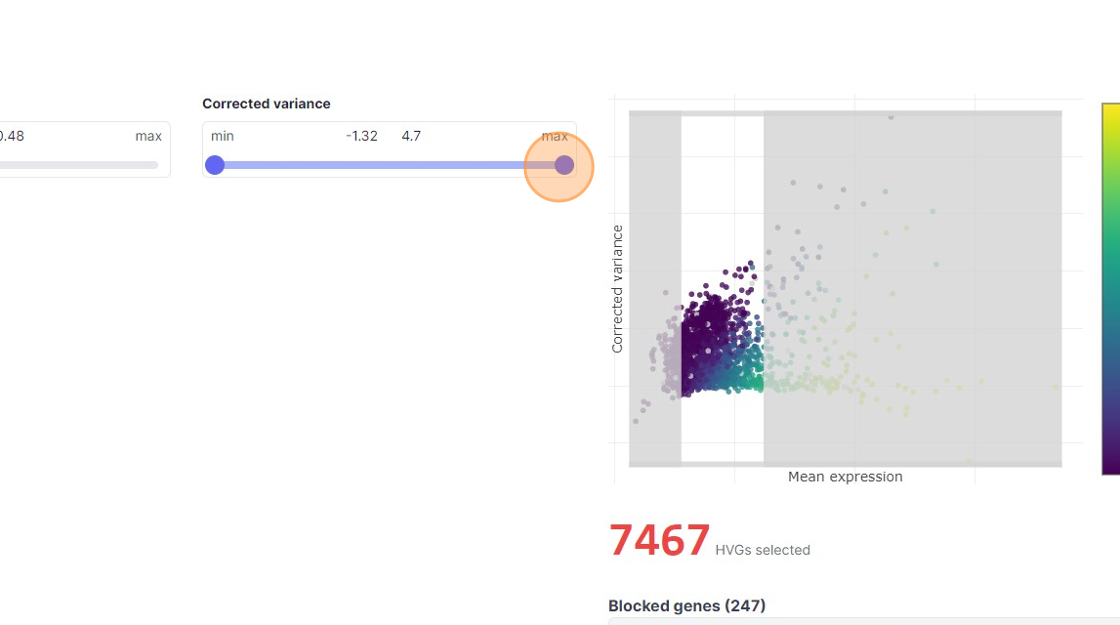
37\. Click here.
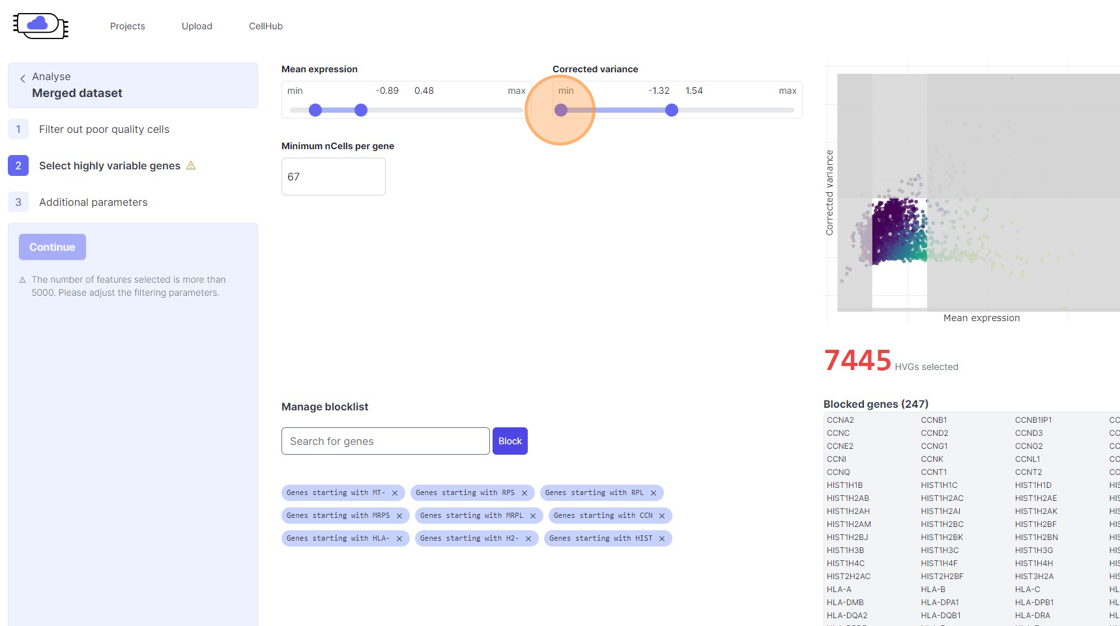
38\. Click "4968"
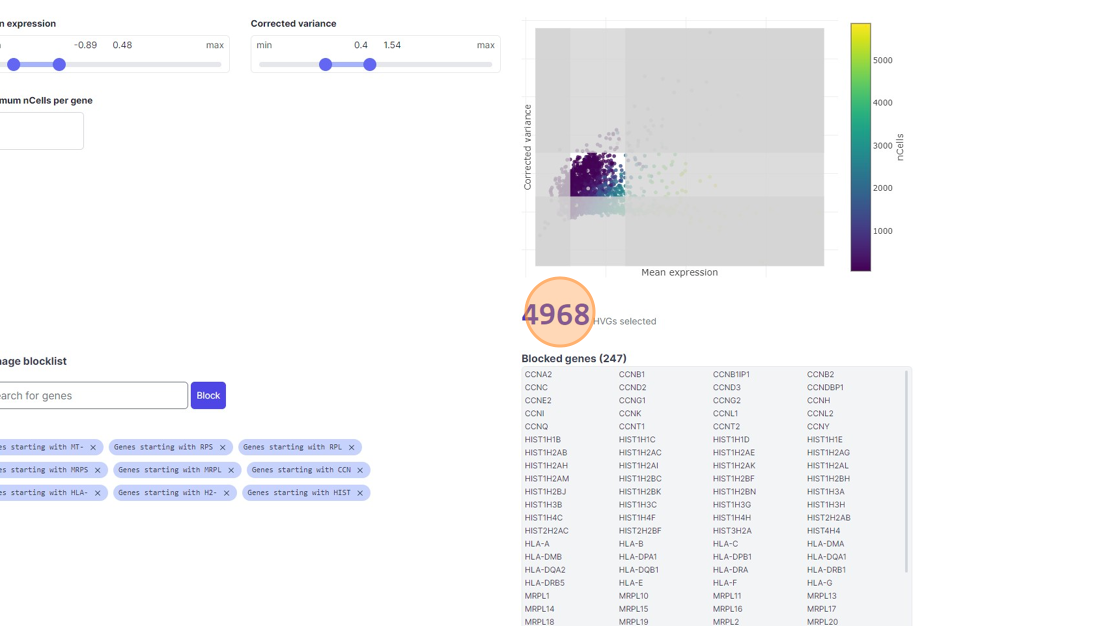
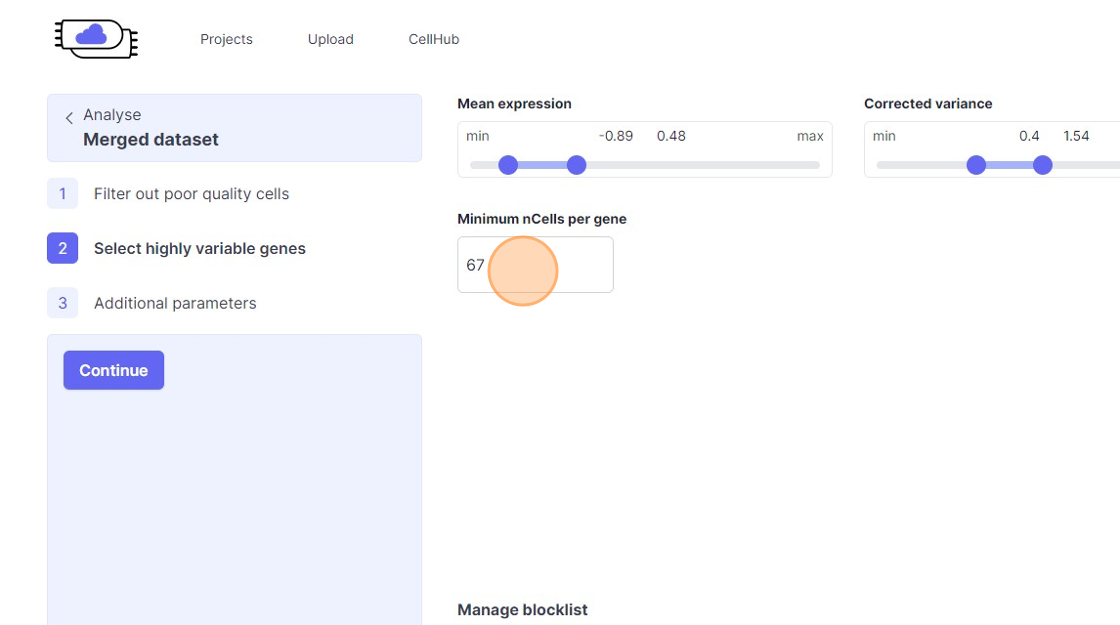
39\. Click the "Minimum nCells per gene" field.
40\. Click the "Search for genes" field.
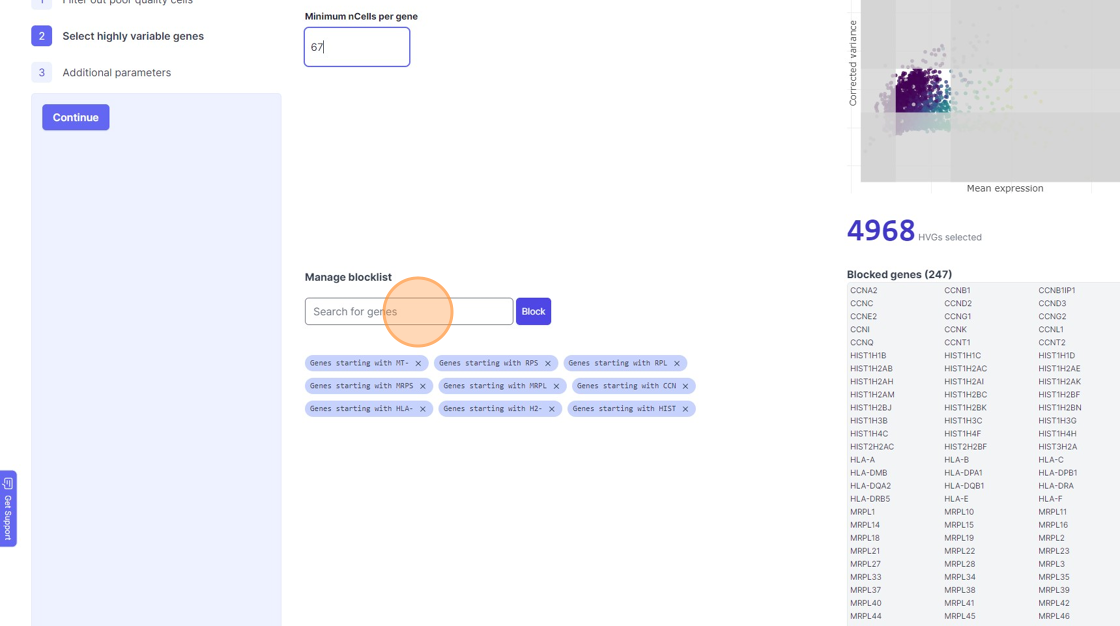
41\. Type "mm"
42\. Click "Found 9 genes starting with MM"
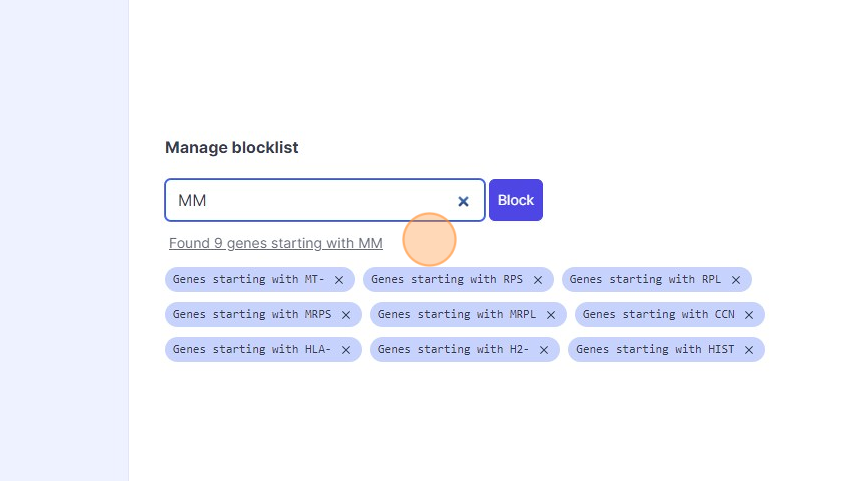
43\. Click "Block"

Additional Analysis Parameters
Batch correction
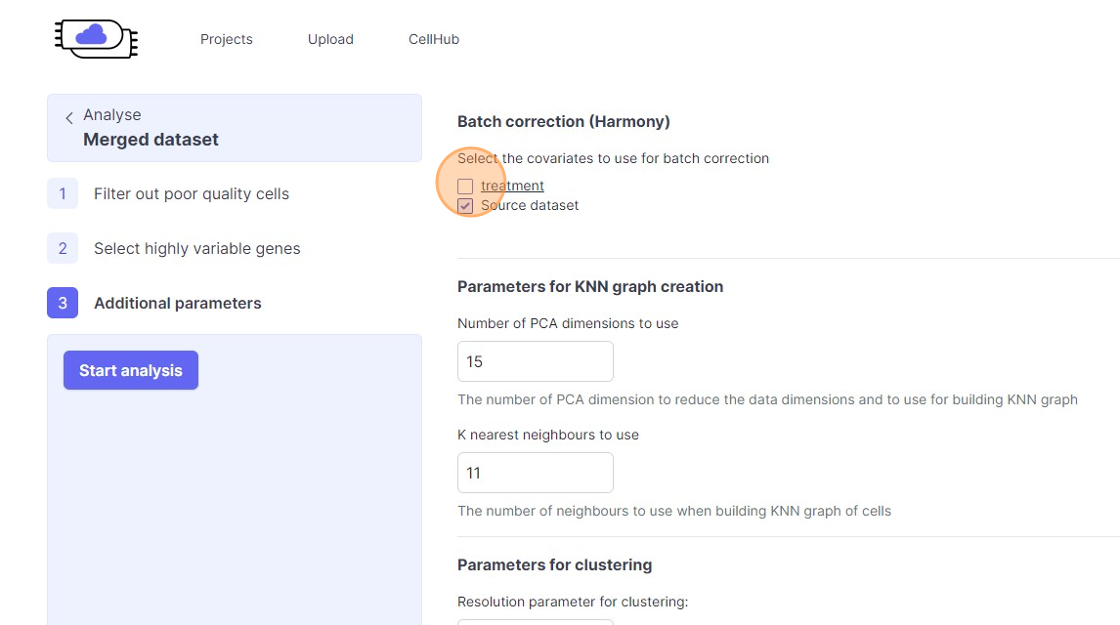
46\. Click the "treatment" field.
47\. Click the "Source dataset" field.
48\. Click "Batch correction will not be used"
49\. Click the "Source dataset" field.
50\. Click the "treatment" field.
51\. Click the "Number of PCA dimensions to useThe number of PCA dimension to reduce the data dimensions and to use for building KNN graph" field.
52\. Click the "K nearest neighbours to useThe number of neighbours to use when building KNN graph of cells" field.
53\. Click the "Resolution parameter for clustering:The resolution parameter is used by Leiden clustering to decide the number of clusters" field.
54\. Click "Start analysis"
55\. Click "Redirecting to projects"
56\. Click here.